In the Hamerman Lab, we are interested in understanding how myeloid cells contribute to both productive and pathological immune responses during infection, inflammatory, and autoimmune diseases.
Our research focuses on monocytes and macrophages, and conventional and plasmacytoid dendritic cells, key players in innate immune responses that set the stage for subsequent adaptive immunity.
We are particularly interested in understanding how signaling by Toll-like receptors (TLRs) is regulated in these innate cells and how dysregulated TLR responses contribute to both initiation and propagation of inflammatory and autoimmune diseases, including systemic lupus erythematosus (SLE) and the autoimmune complication macrophage activation syndrome (MAS).
We also have a key interest in monocyte and macrophage development during homeostasis, and how this process changes during inflammation, whether due to infection, inflammatory or autoimmune diseases.
Our research will lead to a better mechanistic understanding of how TLRs and myeloid cells function and will allow for identification of new therapeutic intervention points in inflammatory and autoimmune diseases.

Jessica Hamerman, PhD
Lab Members

Susan Canny, MD, PhD

Izzy Draper

Lucy Li

Minjian Ni, MD, PhD

Susana Orozco, PhD

Natalie Thulin, BA

Hayley Waterman, PhD
Research Projects
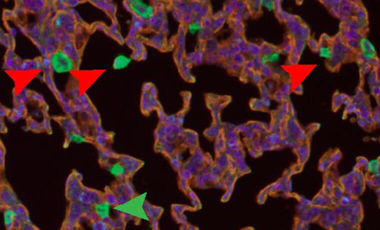
Flightless-1 in lung macrophage and DC development and function
Myeloid cells express a variety of proteins to regulate the actin cytoskeleton, key to migration, adhesion, and phagocytosis.
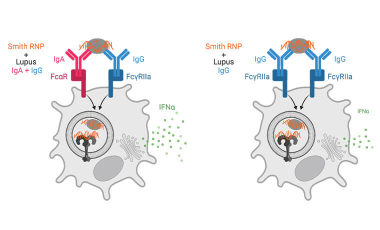
Immune complex activation of pDC IFNα production in lupus
In lupus, Plasmacytoid dendritic cells (pDCs) internalize antibody-containing immune complexes through Fc receptors thereby delivering nucleic acid containing antigens to endosomal TLRs resulting in IFNα production, which amplifies the disease.
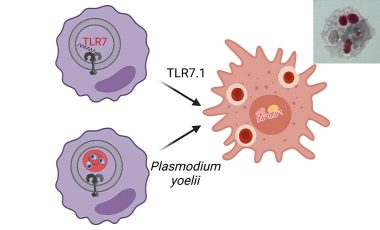
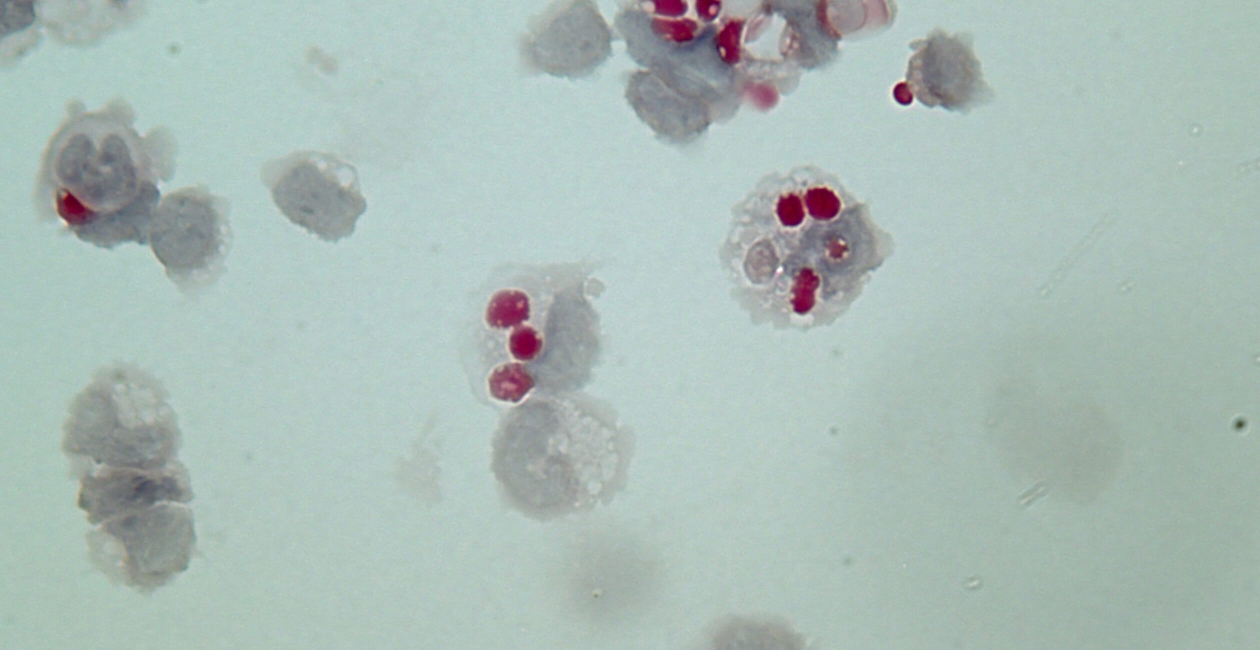
Monocyte-derived inflammatory hemophagocytes in disease
In the Hamerman lab we are working to gain a mechanistic understanding of how monocytes differentiate and contribute to diverse pathologies in autoimmune disease, macrophage activation syndrome and malarial anemia in order to help identify therapeutic targets to restrain diseases.
Featured Publications
-
Identification of biomarkers for COVID-19 associated secondary hemophagocytic lymphohistiocytosis.
bioRxivCanny SP, Stanaway IB, Holton SE, Mitchem M, O'Rourke AR, Pribitzer S, Baxter SK, Wurfel MM, Malhotra U, Buckner JH, Bhatraju PK, Morrell ED, Speake C, Mikacenic C, Hamerman JA -
Lupus IgA1 autoantibodies synergize with IgG to enhance plasmacytoid dendritic cell responses to RNA-containing immune complexes.
Sci Transl MedWaterman HR, Dufort MJ, Posso SE, Ni M, Li LZ, Zhu C, Raj P, Smith KD, Buckner JH, Hamerman JA -
Signals governing monocyte differentiation during inflammation.
Curr Opin ImmunolOrozco SL, Canny SP, Hamerman JA -
Chronic TLR7 and TLR9 signaling drives anemia via differentiation of specialized hemophagocytes.
ScienceAkilesh HM, Buechler MB, Duggan JM, Hahn WO, Matta B, Sun X, Gessay G, Whalen E, Mason M, Presnell SR, Elkon KB, Lacy-Hulbert A, Barnes BJ, Pepper M, Hamerman JA -
Cutting Edge: BCAP Promotes Lupus-like Disease and TLR-Mediated Type I IFN Induction in Plasmacytoid Dendritic Cells.
J ImmunolChu T, Ni M, Chen C, Akilesh S, Hamerman JA

Early-Career Scientist Lucy Li Advances Lupus Research at BRI
Early-career scientists play a vital role in BRI’s work. Meet Lucy Li, who is working to answer pressing questions about lupus as part of her MD–PhD program.

Computational Skills Grant Fuels Collaboration and Discovery
Learn how Susana Orozco, PhD, Hannah DeBerg, PhD, and Jessica Hamerman, PhD, are working together to apply the latest tools to long-standing research questions.

Innovation Fund Spotlight: Single-Cell CRISPR Screening To Explore Genetics and Autoimmune Diseases
Together, Jessica Hamerman, PhD, and John Ray, PhD, are exploring the genetic roots of autoimmune disease using single-cell CRISPR screening.
News

Discovering immune mechanisms that contribute to progressive pulmonary fibrosis
Learn More ➡
Benaroya Research Institute Awarded More Than $14 Million Across 11 Grants To Help Predict, Prevent, Reverse and Cure Immune System Diseases
Read More

